





杨 楠 韩连书 叶 军 邱文娟 张惠文 高晓岚 王 瑜 李筱燕 许 浩 顾学范
内分泌遗传代谢研究室 (上海 200092)
方法 回顾性分析2003年2月—2012年5月就诊的2 547例疑似遗传性代谢病新生儿的血串联质谱和尿气相色谱检测结果,并对其中确诊的127例新生儿的疾病谱及临床表现进行分析。结果 在2 547例新生儿中诊断氨基酸、有机酸和脂肪酸氧化代谢病127例 (5.0%)。其中氨基酸代谢病37例 (29.1%),以鸟氨酸氨甲酰基转移酶缺乏症 (16例) 和枫糖尿病 (14例) 多见;有机酸代谢病76例 (59.8%),以甲基丙二酸血症 (50例) 最常见,其次为丙酸血症 (13例) 和异戊酸血症 (10例);脂肪酸氧化代谢病14例 (11.0%),以多种酰基辅酶A脱氢酶缺乏症多见 (6例)。结论 新生儿期甲基丙二酸血症最常见,其次为鸟氨酸氨甲酰基转移酶缺乏症、枫糖尿病、丙酸血症和异戊酸血症。儿科医师尤其是新生儿科医师应重视利用串联质谱和气相质谱技术进行遗传代谢病检测,以达到早诊断、早治疗的目的。
[临床儿科杂志,2012,30(9):805-808]
中图分类号: R722 文献标志码: A 文章编号: 1000-3606(2012)09-0805-04
Abstract: Objective To investigate the spectrum of neonatal-onset metabolic diseases such as amino acid diseases, organic acidemias and fatty acid oxidation disorders, so as to help pediatricians improve the awareness of these diseases. Methods From February 2003 to May 2012, 2 547 neonates with suspected inherited metabolic disorders from all over China were tested by tandem mass spectrometry (MS/MS) and gas chromatography mass spectrometry (GC-MS). In the 127 confirmed cases, the analysis of disease spectrum and clinical manifestations was performed. Results In the 2 547 cases, a total of 127 cases (5%) were diagnosed with metabolic diseases. Thirty-seven cases (29.1%) were amino acid metabolic diseases in which ornithine transcarbamylase deficiency (16 cases) and maple syrup urine disease (14 cases) were common. Seventy-six cases (59.8%) were organic acid metabolic diseases in which methylmalonic acidemia (50 cases) was the most common, followed by propionic acidemia (13 cases) and isovaleric acidemia (10 cases). Another 14 cases (11%) were fatty acid oxidation disorders in which multiple CoA carboxylase deficiency (6 cases) was common. Conclusions In neonatal period, methylmalonic acidemia is the most common disease, followed by ornithine transcarbamylase deficiency, maple syrup urine disease, propionic acidemia and isovaleric academia. Pediatricians, especially neonatologists, should recognize the importance of the utilization of MS/MS and GC-MS in detection of inherited metabolic disorders and thus achive early diagnosis and early treatment.
Key words: inherited metabolic disorders; tandem mass spectrometry; neonate
遗传性代谢病 (inherited metabolic disorders, MD) 或称先天性代谢缺陷病 (inborn errors of meta-bolism,IEM),是由于遗传性代谢途径缺陷,导致异常代谢物质蓄积或必需物质缺乏,而出现相应的临床症状和实验室检查异常的一类疾病,包括氨基酸、有机酸、脂肪酸、糖和激素等多种物质的代谢异常,目前已报道的疾病种类多达500余种。虽然单一病种患病率低,但因种类繁多,总体发病率约在1/2555~1/784[1]。其中部分疾病在新生儿期发病,临床表现无特异性,诊断困难。近几年开展的血串联质谱和气相质谱技术可检测40余种氨基酸、有机酸和脂肪酸氧化代谢病[2]。本研究室于2002年引进串联质谱技术对氨基酸、有机酸和脂肪酸代谢紊乱等遗传代谢病进行临床研究[3]。本研究分析2003年2月至2012年5月应用串联质谱仪和气相色谱-质谱仪检测并确诊的127例新生儿的氨基酸、有机酸和脂肪酸氧化代谢病的疾病种类,探讨我国新生儿期此类疾病的疾病谱,以提高对氨基酸、有机酸和脂肪酸氧化代谢病的认识,做到早期诊断、早期治疗。
1 对象与方法
1.1 对象
2003年2月至2012年5月,有2547例来自全国各地100余所儿童医院或三级甲等医院儿科(包括澳门仁伯爵和镜湖医院),临床表现疑似遗传性代谢病的新生儿,在本研究室经串联质谱和气相色谱-质谱检测并获得检测结果。其中男1647例、女900例,年龄1~30d。标本采集及检测均经过本院伦理委员会批准并取得家长的知情同意。
1.2 方法
1.2.1 串联质谱检测方法 经足跟采集新生儿血液,滴于专用采血滤纸上,制成干血滤纸片。干血滤纸片经含氨基酸和酰基肉碱内标的甲醇萃取,盐酸正丁醇衍生后,利用串联质谱仪检测酰基肉碱和氨基酸[3,4]。
1.2.2 气相色谱-质谱检测方法 新鲜尿样经尿素酶、盐酸羟铵、氢氧化钠和盐酸处理,除去尿素及蛋白质,并加入17烷酸作为内标,用乙酸乙酯2次萃取,再用双(三甲基硅烷基)三氟乙酰胺与三甲基氯硅烷混合物进行甲基硅烷化衍生后,进行气相色谱-质谱尿有机酸检测[5]。
1.2.3 疾病诊断 疾病的诊断主要依据各种疾病的发病机制,由串联质谱和气相色谱-质谱检测的相应特异指标,参考文献报道进行判断[6,7],结合临床表现、常规实验室检查及相应的治疗效果进行综合诊断,部分患儿经过基因突变检测确诊。
2 结果
2.1 遗传代谢病患儿检测结果及临床资料分析
在2547例新生儿中,诊断127例(5.0%)共17种遗传代谢病,男84例、女43例,年龄中位数9d(1~30d)。其中氨基酸代谢病7种37例(29.1%),有机酸血症5种76例(59.8%),脂肪酸氧化代谢病5种14例(11.0%)。其中17例新生儿在出生3d内的血标本已出现异常,并随后出现临床症状。常见临床表现为:喂养困难87例(68.5%)、抽搐58例(45.7%)、肌张力低下47例(37.0%)。除上述异常外,部分疾病具有相对特异性表现。
2.2 氨基酸代谢病患儿临床特点及特异检测指标
37例氨基酸代谢病患儿临床表现及主要异常指标见表1。鸟氨酸氨甲酰转移酶缺乏症最多见,其次为枫糖尿病和瓜氨酸血症-I型。鸟氨酸氨甲酰转移酶缺乏症表现为高血氨(6例>500μmol/L)、昏迷,血气分析多正常;5例死亡。血串联质谱示瓜氨酸降低,尿气相质谱示乳清酸和尿嘧啶升高。枫糖尿病临床症状重,多为肌张力增高、角弓反张、双下肢“剪刀样”交叉,伴酸中毒,头颅MRI示脑损伤,6例死亡。希特林蛋白缺乏症表现皮肤黄染和肝肿大,肝功能异常,总胆红素和直接胆红素升高,甲胎蛋白显著升高,经无乳糖奶粉治疗患儿恢复正常。
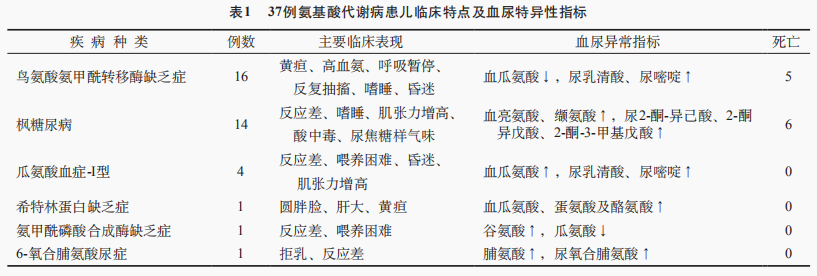
2.3 有机酸血症患儿临床特点及特异检测指标
76例有机酸血症患儿中以甲基丙二酸血症(MMA)最多见,其次为丙酸血症(PA)和异戊酸血症。有机酸血症患儿以酸中毒和神经系统损害表现为主,其中酸中毒49例(64.5%),高乳酸血症14例(18.4%),尿酮体阳性8例(10.5%)。MMA和PA患儿临床多表现为喂养困难、难治性酸中毒、嗜睡、抽搐等,二者临床表现无明显差异,病情危重。本组MMA患儿死亡15例(30.0%),PA患儿死亡2例(15.4%)。串联质谱检测MMA和PA均表现为丙酰肉碱增高。另外,PA患儿伴甘氨酸增高、尿3-羟基丙酸及甲基枸橼酸增高,MMA患儿尿中甲基丙二酸及甲基枸橼酸升高。异戊酸血症患儿10例,多出现抽搐、肌张力异常、特殊汗脚味,死亡5例(50.0%)。戊二酸血症-I型患儿表现为低血糖、酸中毒、脑水肿。有机酸血症患儿的临床特点及血尿检测指标见表2。
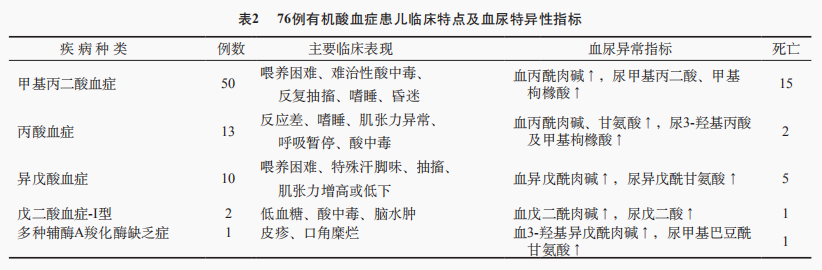
2.4 脂肪酸氧化代谢病患儿临床特点及特异检测指标
脂肪酸氧化代谢病相对较氨基酸或有机酸代谢病少见,共计14例(11.0%),其中多种酰基辅酶A脱氢酶缺乏症6例,短链酰基辅酶A脱氢酶缺乏症3例,原发性肉碱缺乏症2例,极长链酰基辅酶A脱氢酶缺乏症(VLCAD)2例,肉碱棕榈酰转移酶II缺乏症1例。脂肪酸氧化代谢病患儿主要表现为进行性肌张力减退、嗜睡或昏迷等,实验室检查肌酸激酶增高为其特点。尿气相色谱-质谱有机酸检测短链酰基辅酶A脱氢酶缺乏症患儿显示尿乙基丙二酸增高,其他脂肪酸代谢病患儿表现为多种二羧酸增高。短链酰基辅酶A脱氢酶缺乏症以血丁酰肉碱增高为主,多种酰基辅酶A脱氢酶缺乏症以辛酰肉碱增高为主,VLCAD以血肉豆蔻烯酰肉碱和肉豆蔻酰肉碱增高为主,肉碱棕榈酰转移酶II缺乏症以血棕榈酰肉碱增高为主。2例患儿猝死,可能与心脏受累有关。
3. 讨论
新生儿期发病的遗传代谢病发病急,进展快,由于无特异性临床表现,多被误诊为脓毒症、缺氧缺血性脑病等,治疗效果不佳。因此在临床工作中,对所有的危重新生儿,都要警惕遗传代谢病的可能。在新生儿期发病的遗传代谢病,多为氨基酸、有机酸和脂肪酸氧化代谢病。近年来,随着串联质谱仪的应用,遗传代谢病患儿检出率较前明显提高。Zytkovicz等[8]研究表明,50%的遗传代谢病确诊病例来源于只占人口总数5%的NICU人群,说明NICU人群是遗传代谢病发生的高危人群。本研究在2547例遗传代谢病疑似新生儿中检出氨基酸、有机酸和脂肪氧化代谢病患儿127例,检出率为5.0%,远高于普通人群。其中又以有机酸血症检出率最高,占59.8%;其次为氨基酸代谢病37例,占29.1%;而脂肪酸氧化代谢病较少见,占11.0%。本研究结果与香港地区新生儿筛查发现氨基酸代谢病、有机酸代谢病和脂肪酸代谢病的患病率之比约为5:1:1有所差异[9],原因可能是香港地区为经新生儿筛查确诊患儿,而本研究为新生儿期发病患儿,提示部分氨基酸代谢病在新生儿期并没有发病。另外,脂肪酸氧化代谢病在欧美国家患病率较高,以中链酰基辅酶A脱氢酶缺乏症(MCAD)最多见[10],与本研究不同。
有机酸代谢障碍是由于氨基酸、脂肪及糖代谢途径中某种酶的缺陷,导致其中间代谢产物有机酸增加,有机酸过多蓄积造成神经系统及其他脏器损害,严重时出现酸中毒、低血糖和高血氨等。甲基丙二酸血症在有机酸代谢障碍中最常见,其次是丙酸血症和异戊酸血症。国外大样本筛查结果表明,甲基丙二酸血症发病率在1/169000~1/48000之间[11]。本研究中,甲基丙二酸血症患儿占有机酸血症的65.8%,表明在中国甲基丙二酸血症是最常见的有机酸血症。甲基丙二酸血症主要表现为嗜睡、反应差、抽搐等神经系统症状,实验室检查常提示代谢性酸中毒、高血氨,病情进展迅速,短期内出现代谢危象,患儿昏迷,甚至死亡。本研究中死亡15例(30%),进一步证实新生儿期发病的甲基丙二酸血症患者病情危重,如未能早期诊断,及时治疗,可引起严重后遗症或死亡。
氨基酸代谢病是由于氨基酸代谢障碍所造成的一组疾病。在我国新生儿筛查疾病中苯丙酮尿症患病率最高,达1:11144[12]。本研究结果显示,在新生儿期发病的氨基酸代谢病中,鸟氨酸氨甲酰基转移酶缺乏症最常见,占43.2%;枫糖尿病在新生儿期也较常见,在本研究中占氨基酸代谢病的37.8%。
国外近几年利用串联质谱检测血酰基肉碱水平,用于脂肪酸氧化代谢病的新生儿筛查和临床高危患儿的诊断,发现脂肪酸氧化代谢病的患病率较以往增高。本研究结果显示,新生儿期发病的脂肪酸代谢病较氨基酸代谢病、有机酸血症少,且多为多种酰基辅酶A脱氢酶缺乏症。脂肪酸氧化代谢病患儿临床表现多样化,无特异性,主要影响肝脏、心脏和骨骼肌。Dott等[13]对不明原因死亡的793例患儿的干血滤纸片进行串联质谱检测,诊断4例(0.5%)脂肪酸氧化代谢病,2例MCAD,2例VLCAD,提示猝死的病因之一为脂肪酸氧化代谢病。综上所述,当新生儿期遇到原因不明的反应差、喂养困难、抽搐、意识障碍和肌张力异常等神经系统表现,或有呕吐、喂养困难和肝脾肿大等多脏器损害的症状体征,常规实验室检查有酸中毒、高血氨、高乳酸血症、低血糖、肝功能异常、贫血和酮症,或头颅CT、MRI表现为不同形式的脑损害时,应考虑到有机酸、氨基酸和脂肪酸氧化代谢病的可能,及早留取血样或尿样进行血串联质谱、尿气相质谱特异性检测,可达到早诊断、早治疗的目的,有利于降低新生儿期死亡率,减轻或避免不可逆的脏器损伤。
参考文献:
[1] Therrell BL,Adams J. Newborn screening in North Ameri- can [J]. J Inherit Metab Dis,2007,30(4):447-465.
[2] [2] Frazier DM,Millington DS,McCandless SE,et al. The tandem mass spectrometry newborn screening experience in North Carolina: 1997–2005 [J]. J Inherit Metab Dis, 2006,29(1):76–85.
[3] 韩连书,高晓岚,叶军,等. 串联质谱技术在有机酸 血症鉴别诊断中的应用 [J]. 临床儿科杂志,2006, 24(12):973-977.
[4] 韩连书,叶军,邱文娟,等. 串联质谱联合气相色谱- 质谱检测遗传性代谢病 [J]. 中华医学杂志,2008, 88(30):2122-2126.
[5] 罗小平,王慕逖,魏虹,等. 尿滤纸片法气相色谱-质谱 分析技术在遗传性代谢病高危筛查诊断中的应用 [J]. 中 华儿科杂志,2003,41(4):245-248.
[6] Garg U,Dasouki M. Expanded newborn screening of in- herited metabolic disorders by tandem mass spectrometry: clinical and laboratory aspects [J]. Clin Biochem,2006, 39(4):315-332.
[7] Schulze A,Lindner M,Kohlmuller D,et al. Expanded newborn screening for inborn errors of metabolism by elec- trospray ionization-tandem mass spectrometry: results, out- come, and implications [J]. Pediatrics,2003,111(6 Pt 1): 1399-1406.
[8] Zytkovicz TH,Fitzgerald EF,Marsden D,et a1. Tam- dem mass spectrometric analysis for amino, organic,and fatty acid disorders in neonatal dried b1ood spots: a two- year summary from the New England Neonatal Screening Program [J]. Clin Chem. 2001,47(11):1945-1955.
[9] Lee HC,Mak CM,Lam CW,et al. Analysis of inborn er- rors of metabolism: disease spectrum for expanded newborn screening in Hong Kong [J]. Chin Med J (Engl), 2011, 124(7):983-989.
[10] Grosse SD,Khoury MJ,Greene CL,et al. The epidemi- ology of medium chain acyl-CoA dehydrogenase deficiency- an update [J]. Genet Med,2006,8(4):205–212.
[11] 王斐,韩连书. 甲基丙二酸血症诊治研究进展 [J]. 临床儿 科杂志,2008,26(8):724-727.
[12] 顾学范,王治国. 中国580万新生儿苯丙酮尿症和甲状腺 功能减低症筛查 [J]. 中华预防医学杂志,2004,38(2): 99-102.
[13] Dott M,Chace D,Fierro M,et al. Metabolic disorders detectable by tandem mass spectrometry and unexpected early childhood mortality: a population-based study [J]. Am J Med Genet A,2006,140(8):837–842.







